
Repurposed Drug Extends Life of Kids with Progeria
/The ultra-rare genetic condition was uniformly fatal. That may be changing now.
Let me say up front that I am NOT talking about a randomized trial this week. Nevertheless, this study appearing in the Journal of the American Medical Association has data that is more compelling than many trials that have come across my desk.


Survival among individuals with progeria

Lonafarnib - a farnesyl-transferase inhibitor
The disease is Hutchinson-Gilford Progeria, an ultra-rare condition – occurring in just 1 in 4 million births and characterized by premature aging.
The natural history of progeria is death, usually due to cardiac causes, in the early to mid-teen years. Until recently, there have been no known therapies.
Enter lonafarnib, an obscure little farnesyl transferase inhibitor that has seen some use in hepatitis delta virus infection and is being investigated for its anti-cancer properties.
The primary gene defect in progeria is in lamin A. Lamin A normally gets farnesylated, but the farnesyl part of the protein is subsequently cleaved. In progeria, that cleavage site is rendered inactive, leading to retention of the farnesyl group and accumulation of the altered protein which leads to the progeria phenotype.
Researchers, led by Leslie Gordon, wondered whether inhibiting farnesylation in the first place would make a difference. Lonafarnib does just that.
The first trial of lonafarnib led to promising results – kids treated with the drug had improved weight gain and less skeletal rigidity. But only now was there enough data collected to look at a hard outcome – all-cause mortality.
The researchers compared 63 patients treated with lonafarnib to 63 patients who were not treated with the drug. Again, this wasn’t randomized. I think there’s a good argument here that randomization would be unethical. That said, the researchers matched treated and untreated patients on a variety of factors to minimize bias. And the results were dramatic.
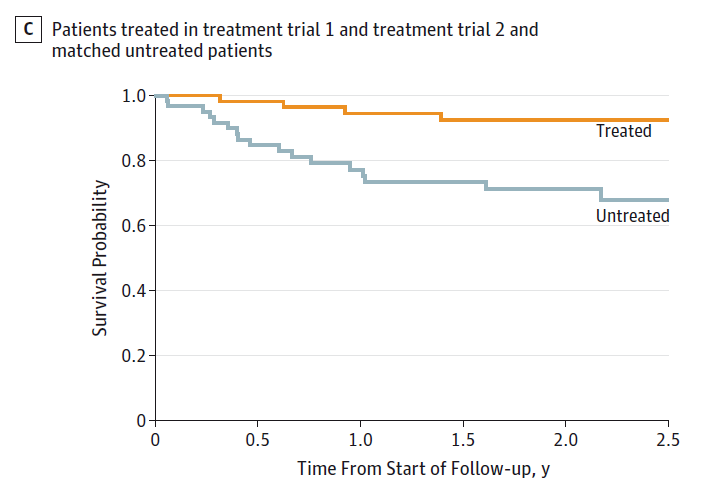
The survival curves show what the raw numbers tell us. Among the treated patients, just 4 died over the two years of study, compared to 17 deaths in the untreated group. These results are unheard of in this disease. Results this strong are rarely seen in most diseases, actually.
Now most of these kids started taking the medication at around 9 or 10 years of age, and it didn’t seem to have much effect on their physical appearance. But one wonders if earlier treatment would lead to even more dramatic results.
This is a story that is inspiring on many levels. There’s, obviously, the potential to transform the lives of a group of very special children. But it’s also exemplary of the scientific method gone right – where rigorous basic science and the smart application of an existing drug combine to produce outcomes that are genuine breakthroughs.